Polycythemia vera, classified as a myeloproliferative neoplasm (MPN) and characterized by uncontrolled, clonal, myeloid expansion with predominant erythrocytosis,1 affects about 100,000 individuals in the United States.2 It is a chronic and burdensome disease associated with shortened survival.3 Patients are at an increased risk of cardiovascular events, solid tumors, and transformation to myelofibrosis (MF) and/or acute myeloid leukemia (AML).4,5 Furthermore, patients generally have a reduced quality of life (QoL) stemming from prevalent and occasionally severe polycythemia vera–related signs and symptoms, including fatigue, pruritus, and splenomegaly.6 In general, the classical Philadelphia chromosome-negative MPNs are associated with driver mutations in the following three genes: Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL).7 Almost all patients with polycythemia vera have an activating mutation in the cytoplasmic signal transduction protein JAK2.4 Patients with essential thrombocythemia (ET) or MF can have mutations in JAK2, CALR, or MPL. However, CALR and MPL mutations are absent or exceedingly rare in patients with polycythemia vera.7 Diagnosis can be challenging and is currently based on 2016 World Health Organization (WHO) diagnostic criteria.1
Management strategies include the use of aspirin, phlebotomy, and cytoreductive therapy. Ruxolitinib is a newer treatment option available for patients with polycythemia vera who are either resistant to or intolerant of hydroxyurea8,9— a population that previously had few treatment options. It is important for community oncologists and other treating clinicians to understand current diagnostic strategy and management options based on established guidelines, recent clinical evidence, and regulatory updates.
Search and selection process for research sources
In September 2016, we searched PubMed for articles published since 2006 with polycythemia vera included in the abstract or title. The initial 1,730 publications were screened by eye to select 46 key articles that guide current management of polycythemia vera. Four studies published before 2006 were also included based on their continued relevance.
Epidemiology and pathophysiology
Based on a meta-analysis of patients from Europe and the United States, the annual incidence of polycythemia vera estimated to be between 0.7 and 2.6 per 100,000 people.10 The age-adjusted prevalence of polycythemia vera in the United States is about 45-57 per 100,000 people,2 however, the true prevalence might be considerably greater.
Patients with polycythemia vera are at increased risk of cardiovascular events, thrombosis, and death.3-5 Risk is highest among patients older than 60 years or with a history of thrombosis.11 Uncontrolled myeloproliferation has also been identified as a risk factor for cardiovascular mortality and thrombosis. This was demonstrated in the prospective Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial, which reported more cardiovascular events in patients with hematocrit levels of 45%-50%, compared with those whose hematocrit levels were <45%.12 In addition, retrospective data suggest leukocytosis is a potential risk factor for thromboembolic events and poor outcomes.13
Dysregulated JAK2 signaling is the principal driver of polycythemia vera pathophysiology. About 95% of patients with polycythemia vera will have an identifiable JAK2 V617F exon 14 mutation, with an additional 3%-5% demonstrating a JAK2 exon 12 mutation.4,14 Under physiologic conditions, JAK2 interacts with the STAT family of signal transduction proteins and serves as an important regulator of normal hematopoiesis.15 Mutated, constitutively activated JAK2 signaling promotes the various polycythemia vera disease manifestations, including excessive myeloproliferation, splenomegaly,15 and constitutional symptoms.14,16,17
Burden of disease for the individual
Mortality
Patients with polycythemia vera have an increased risk of mortality compared with an unaffected, age- and gender-matched cohort of the general population.3 A retrospective study of Medicare patients with polycythemia vera (mean age at diagnosis, 76.1 years) reported a median survival of 5.4 years, compared with 8.7 years for a matched cohort.3 A second retrospective study reported a median survival of 13.5 years (median age at diagnosis, 64 years; median follow-up time, 11.8 years).18
Leading causes of death for patients with polycythemia vera include cardiovascular and thrombotic events, the development of secondary solid tumors, and disease transformation to MF and/or AML. In the prospective European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) study of 1,638 patients, 45% of deaths (74/164) resulted from cardiovascular causes (1.7 per 100 patient-years).5 Thirteen percent of deaths were related to either leukemic or myelofibrotic transformation, and 20% of deaths were attributed to secondary solid tumors.5 In a retrospective analysis of 1,545 patients with polycythemia vera followed for a median of 6.9 years after diagnosis, 347 had died, primarily from acute leukemia (10%), secondary malignancies (10%), and thrombotic events (9%).4 Arterial and venous thrombotic events occurred in 12% and 9% of patients, respectively, with disease transformation to MF and AML occurring in 9% and 3% of patients. Further support of an increased risk of secondary malignancies comes from a retrospective analysis of a large Swedish cancer registry (1958–2006) that found an increased risk of secondary endocrine, renal, and skin malignancies; MF; and leukemia among patients with polycythemia vera.19
Symptoms and quality of life
Symptoms of polycythemia vera vary in severity, and patients often fail to attribute symptoms to the disease.20 Moreover, clinicians may underestimate a patient’s true disease burden or the effect it has on QoL.20 Point-of-care metrics, such as the MPN Symptom Assessment Form (MPN-SAF), were developed to aid in identifying and grading symptom burden. Studies using this metric have reported fatigue as the most common and most severe symptom (incidence, 73%-92%), with a variety of other symptoms also affecting a majority of patients (Figure 1).6,21-24 Although fatigue, pruritus, and a higher MPN-SAF total symptom score are significantly correlated with reduced QoL,22,25 the recent MPN Landmark survey suggests that even patients with low symptom severity scores have a reduction in their QoL.6 This study also highlighted that polycythemia vera can adversely affect multiple aspects of daily living: 48% of patients reported disease interfering with daily activities; 63% with family or social life; and 37% with employment, feeling compelled to work reduced hours.6
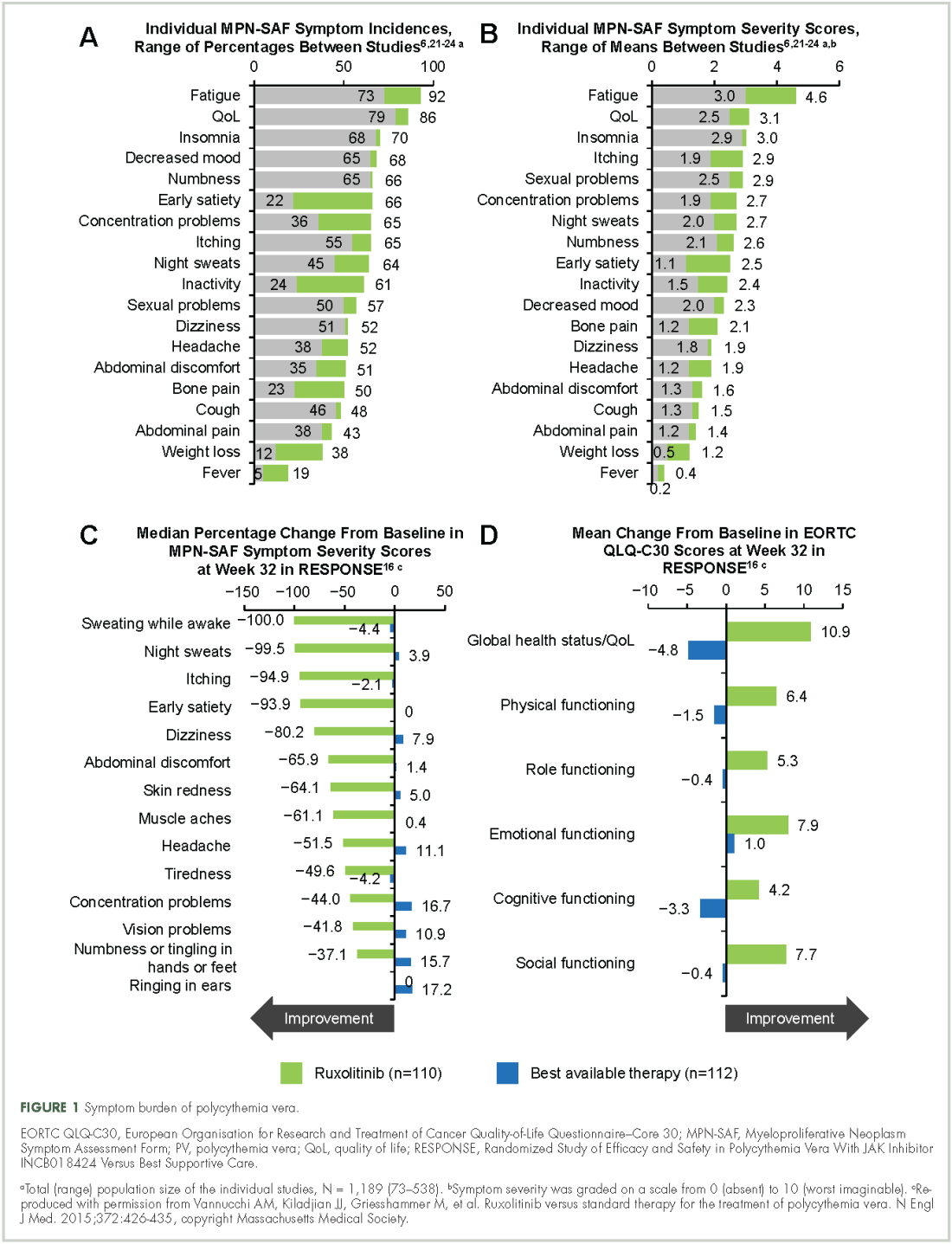
Splenomegaly is a common feature of polycythemia vera, affecting an estimated one in three patients, which may result in discomfort and early satiety.4
Identification and diagnosis
Most patients diagnosed with polycythemia vera are between the ages of 60 and 76 years,3-5 although about 25% are diagnosed before age 50.4 Tefferi and colleagues reported in a retrospective study that common features at presentation include JAK2 mutations (98%), elevated hemoglobin (73%), endogenous erythroid colony growth (73%), white blood cell count of >10.5 × 109/L (49%), and platelet count of ≥450 × 109/L (53%).4 In that same study, about a third of patients presented with a palpable spleen or polycythemia vera–related symptoms, including pruritus and vasomotor symptoms. However, many patients were asymptomatic at presentation, diagnosed incidentally by abnormal laboratory values.4 Patients can present with vascular thrombosis, occasionally involving atypical sites (eg, Budd-Chiari syndrome, other abdominal blood clots),26 thus, a heightened awareness and testing for JAK2 mutations may be appropriate in the evaluation of such individuals.
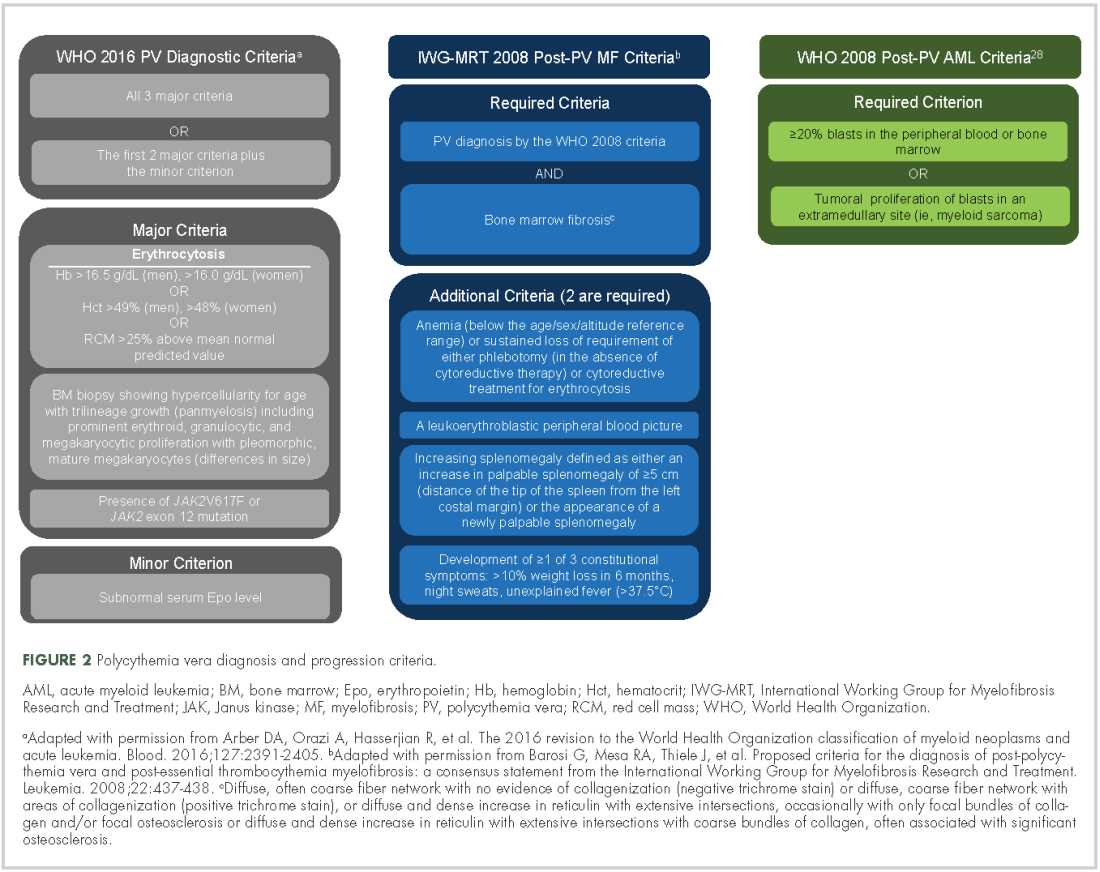
Evidence suggests many clinicians may not rigidly apply the WHO diagnostic criteria to establish a diagnosis.1,27,28 A recent retrospective claims analysis showed that only 40% of 121 patients diagnosed with polycythemia vera met the 2008 WHO diagnostic criteria, and for some patients, the diagnosis was based solely on the presence of the JAK2 V617F mutation.29 One should be aware of individuals with “masked” polycythemia vera, who may present with characteristic polycythemia vera features but have hemoglobin levels below those established by the WHO in 2008, typically owing to iron deficiency and/or a disproportionate expansion of plasma volume.30 To improve polycythemia vera diagnosis, the WHO diagnostic criteria were updated in 2016 with reduced hemoglobin diagnostic thresholds (Figure 2).1
Management strategy
Treatment goals
The primary polycythemia vera–treatment goals are to reduce the risk of cardiovascular, thrombotic, and hemorrhagic events; reduce the risk of fibrotic and/or leukemic transformation; and alleviate polycythemia vera–related symptoms.11,31
Traditional treatment options
Aspirin. To reduce the risk of death from cardiovascular events, patients with polycythemia vera should receive low-dose aspirin32 and undergo phlebotomy to maintain a target hematocrit <45%, as established by the ECLAP and CYTO-PV trials (Figure 3).4,5,12,13,16,32-36 Higher doses of aspirin (ie, 325 mg 2 or more times a week) are associated with a dose-dependent increased risk of gastrointestinal bleeding.37 Low-dose aspirin is generally well tolerated; however, patients with extreme thrombocytosis may develop bleeding as a consequence of a well-described, thrombocytosis-associated acquired von Willebrand disease.11,38
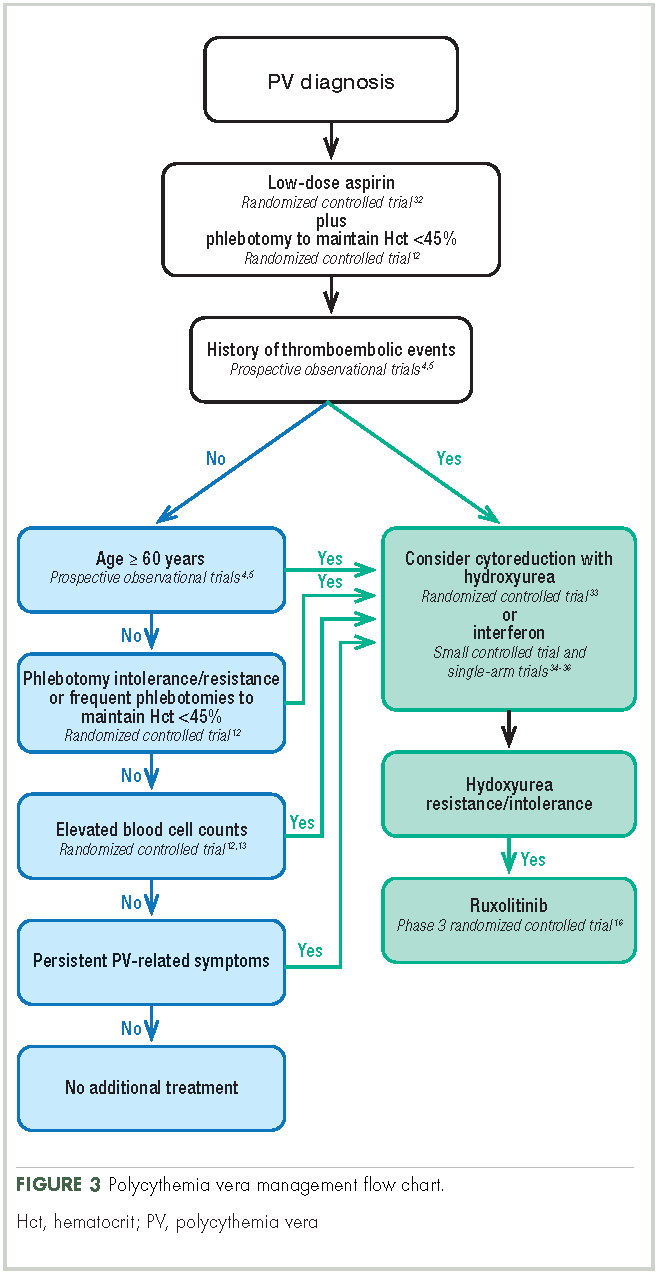
Phlebotomy. This procedure is generally tolerated by most patients, although it can occasionally engender extreme anxiety in some patients39 and may promote clinical manifestations of iron deficiency, including restless leg syndrome,40 impaired cognition, and worsening of fatigue.41 In the CYTO-PV study, 28% of patients with a target hematocrit 45%-50% discontinued phlebotomy treatment, although the percentage that discontinued because of poor tolerance was not reported.12 To avoid potential complications in patients with underlying cardiovascular disease, smaller-volume phlebotomies are often pursued.42
Cytoreductive therapy. Cytoreductive therapy with hydroxyurea or interferon (IFN) is recommended for high-risk patients (ie, those with a history of thrombosis or older than 60 years) as well as those with intolerable symptoms, progressive splenomegaly, or a burdensome phlebotomy requirement.11,31 Hydroxyurea is the typical first-line cytoreductive therapy11 based on clinical benefit,33,43 low cost, and feasibility of long-term treatment.33
Most patients benefit from long-term treatment with hydroxyurea; however, 25% develop resistance to or intolerance of hydroxyurea therapy.44 Intolerance typically manifests as leg ulcers or other mucocutaneous toxicity, gastrointestinal side effects, or fever.44 Resistance to hydroxyurea is defined as failure to achieve phlebotomy independence, persistent leukothrombocytosis or splenomegaly despite adequate doses of hydroxyurea, or inability to deliver the drug owing to dose-limiting cytopenias. The European LeukemiaNet (ELN) formally codified and published a definition of hydroxyurea resistance/intolerance45 (Table), which can be used to identify patients at high risk of poor outcomes.44 In a retrospective chart review of 261 patients with polycythemia vera, those meeting the ELN definition of hydroxyurea resistance had a 5.6-fold greater risk of mortality and a 6.8-fold increased risk of fibrotic and/or leukemic disease transformation.44

The use of IFN-α and pegylated variants are associated with clinical benefit, including normalization of blood counts, reduction of splenomegaly, symptom mitigation, and reduction in JAK2 V617F allele burden.46 However, poor tolerance46 and an inconvenient route of administration often preclude the long-term use of these agents. Adverse events associated with IFN-α include chills, depression, diarrhea, fatigue, fever, headache, musculoskeletal pain, myalgia, nausea, and weight loss.46 In clinical trials, recombinant IFN-α discontinuation rates within the first year of administration were as high as 29% and may have been dose dependent.46
Traditional treatment options may not effectively alleviate polycythemia vera–related symptoms.23,47 Two prospective studies failed to show an improvement in patient-reported MPN-SAF scores after treatment with hydroxyurea, aspirin, phlebotomy, IFN-α, busulfan, or radiophosphorus,23,47 and symptoms may worsen with the use of IFN-α.47
Allogenic transplantation. Although allogeneic transplantation is a potentially curative treatment option, it has been reserved primarily for younger patients with MPNs (age <60 years31). Furthermore, a recent systematic review concluded that overall survival was worse following allogeneic transplantation compared with a nontransplant approach (ie, phlebotomy and aspirin).48
Ruxolitinib. The oral JAK1/JAK2 inhibitor ruxolitinib has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea,8 and by the European Medicines Agency (EMA) for adult patients with polycythemia vera who are resistant to or intolerant of hydroxyurea.9 Ruxolitinib is also approved by the FDA for patients with intermediate- or high-risk MF, including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF,8 and for similar patient populations by the EMA.9
Approval of ruxolitinib for the treatment of patients with polycythemia vera was based on the phase 3 randomized, open-label, multicenter RESPONSE trial in which 222 patients with polycythemia vera who met the modified ELN criteria for hydroxyurea resistance or intolerance (Table)16,31 were randomized to ruxolitinib or best available therapy (BAT). Compared with BAT, a greater proportion of patients treated with ruxolitinib achieved the primary composite endpoint of hematocrit control without the need for phlebotomy and ≥35% reduction in spleen volume by week 32 (22.7% vs 0.9%; P < .001).16,49 When looked at individually, hematocrit control and reduction in spleen size favored ruxolitinib over BAT (hematocrit control, 60.0% vs 18.8%, ruxolitinib and BAT, respectively; ≥35% reduction in spleen volume, 40.0% vs 0.9%). Furthermore, more patients receiving ruxolitinib achieved the key secondary endpoint of complete hematologic remission than did those receiving BAT (ie, normalization of blood counts; 23.6% vs 8.0%; P = .0016).16,49 Of note is that most patients who achieved primary treatment responses maintained disease control for ≥80 weeks.49
Results from RESPONSE indicate that ruxolitinib may substantially improve polycythemia vera–related symptoms. Treatment with ruxolitinib was associated with a greater improvement in nearly all symptoms evaluated by the MPN-SAF as well as greater improvements in QoL and functional measures with the EORTC QLQ-C30 trial metric compared with BAT (Figure 1).16 In addition, a post hoc exploratory analysis of RESPONSE indicated that patients receiving ruxolitinib showed a rapid normalization of abnormal iron indices at baseline, compared with those receiving BAT.50
Treatment safety and tolerability are particularly important considerations for patients with polycythemia vera, given the long natural history of the disease. In a preplanned analysis of RESPONSE at 80 weeks, 83% of patients randomized to receive treatment with ruxolitinib remained on treatment (median exposure, 111 weeks).49 Most adverse events reported in both treatment arms were grade 1/2.16,49 The most frequent nonhematologic adverse events (per 100 patient-years of exposure) in the ruxolitinib arm were headache (10.5%), diarrhea (9.7%), pruritus (9.7%), and fatigue (8.3%). The most common grade 3/4 nonhematologic adverse events (occurring at a rate of ≥0.9 per 100 patient-years of exposure) were limited to dyspnea (1.3%), abdominal pain (0.9%), headache (0.9%), and herpes zoster (0.9%).49 Hematologic adverse event rates in the ruxolitinib and BAT arms included anemia (any grade, 27.2% vs 47.6%, respectively; grade 3/4, 0.9% vs 0%), lymphopenia (27.2% vs 78.8%; 9.7% vs 27.2%), and thrombocytopenia (14.9% vs 29.9%; 2.6% vs 5.4%).49 Herpes zoster infections occurred more frequently in the ruxolitinib arm (any grade, 5.3%; grade 3/4, 0.9%) compared with the BAT arm (no herpes zoster events).49 There was a higher rate of nonmelanoma skin cancer (NMSC) in the ruxolitinib arm (4.4%), compared with the BAT arm (2.7%),49 most of which occurred in patients with a history of NMSC or precancerous skin lesions.16 Grade 1 or 2 elevations in serum lipids and cholesterol were observed with ruxolitinib but not BAT; however, subsequent effects on patient outcomes have not been determined.8,16 The rates of MF and AML transformations were 1.3% and 0.4%, respectively, in patients randomized to receive ruxolitinib,49 similar to previously published reports for patients with polycythemia vera.44
Additional insight regarding the effect of ruxolitinib on polycythemia vera–related symptoms is available from the RELIEF trial, a randomized, multicenter, double-blind, double-dummy, phase 3b clinical trial. In RELIEF, 110 patients were randomized to receive ruxolitinib or a stable dose of hydroxyurea and were then asked to record disease-related symptoms.51 Although the study failed to meet its primary endpoint (a ≥50% improvement by week 16 in MPN-SAF total symptom score for the cytokine symptom cluster [sum of individual scores for tiredness, itching, muscle aches, night sweats, and sweats while awake]), a numerically greater proportion of patients receiving ruxolitinib achieved the primary endpoint compared with those receiving hydroxyurea (43.4% and 29.6%, respectively; P = .139; odds ratio, 1.82; 95% confidence interval, 0.82-4.04). Similarly, the proportion of patients reporting a ≥50% improvement in pruritus and fatigue favored ruxolitinib over hydroxyurea (itching, 40.0% vs 26.4%; tiredness, 54.2% vs 32.0%). The safety profile for ruxolitinib was similar to that reported in the RESPONSE trial.
Possible future treatment options
Other possible treatment options for patients with polycythemia vera that are currently in clinical development include three pegylated IFN-α (PEG-IFN-α) variants and the telomerase inhibitor, imetelstat.
Pegylated interferon-α. PEG–IFN-α has the advantage of a longer plasma half-life compared with conventional IFN-α, permitting administration once per week or less often.46 Currently, three variants are under active investigation in phase 3 clinical trials: PEG–IFN-α2a (NCT01259856 and NCT01387763), PEG–IFN-α2b (NCT01387763), and AOP2014/P1101 (NCT02218047, NCT02523638, and NCT01949805).
Imetelstat. The telomerase inhibitor imetelstat is in clinical development for patients with MPNs. Clinical benefit was previously observed in patients with primary MF as well as post-polycythemia vera and post-ET MF.52 Imetelstat was evaluated in a phase 2 trial in patients with polycythemia vera or ET who required cytoreductive therapy and were resistant to or intolerant of ≥1 previous line of therapy or who refused standard therapy (NCT01243073). Results in patients with ET were published,53 however, findings from the polycythemia vera cohort have not been reported.
Community oncologist role in managing disease burden
Most patients with polycythemia vera are managed in the community setting. Consequently, the community oncologist plays a critical role in the initial diagnosis, risk stratification, patient education, and disease management.
Early disease recognition allows prompt therapeutic intervention with low-dose aspirin and phlebotomy, interventions shown to reduce the risk of cardiovascular events based on the ECLAP32 and CYTO-PV trials,12 respectively. The diagnosis of polycythemia vera is facilitated by applying the WHO diagnostic criteria (Figure 2);1 however, one should be aware of atypical presentations, including “masked” polycythemia vera,30 as well as the development of thrombosis at atypical sites.26
Optimal management strategies must include the frequent assessment of symptom burden and its effect on a patient’s QoL, with a keen awareness of the nonspecific nature of polycythemia vera–related symptoms, and the potential for patients and clinicians to minimize that effect.20 Patient-reported symptom severity and QoL should be assessed at each office visit with validated instruments, such as the MPN-SAF 10-item questionnaire.22It is important for the community oncologist to define treatment goals and implement a plan that reduces disease-associated morbidity and mortality. A critical treatment goal is to maintain hematocrit <45% by the appropriate use of phlebotomy12 and/or cytoreductive agents.12,16,46 Continued reassessment is important to identify patients with progressive disease and those who fail to achieve stated treatment goals or require an adjustment in cytoreductive therapy. Oncologists should be familiar with the concept of hydroxyurea resistance/intolerance as defined by the ELN (Table)31,45 to allow early identification of those patients who are most likely to benefit from a treatment change for continued optimal outcome.
Conclusions
Polycythemia vera is a clonal myeloproliferative neoplasm associated with significant disease-related morbidity and mortality. Appropriate management includes early diagnosis and implementation of appropriate therapy according to patient risk and therapeutic tolerance. Patients should initially receive aspirin32 and phlebotomy,12 with the goal of maintaining hematocrit <45%. Higher-risk patients and those who had inadequate disease control with phlebotomy alone require cytoreduction, typically with hydroxyurea. Although most patients will achieve adequate disease control with hydroxyurea,33,43 one in four patients will develop drug resistance or intolerance.44 Ruxolitinib is approved by the FDA and the EMA for the treatment of patients with polycythemia vera who are resistant to or intolerant of hydroxyurea. Compared with BAT, ruxolitinib is associated with improved hematocrit control, reductions in spleen size, a greater probability of blood count normalization, and improvement in polycythemia vera–related symptoms.16
Acknowledgments
Writing assistance was provided by Cory Pfeiffenberger, PhD, of Complete Healthcare Communications LLC, and was funded by Incyte Corporation, the maker of ruxolitinib.