Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
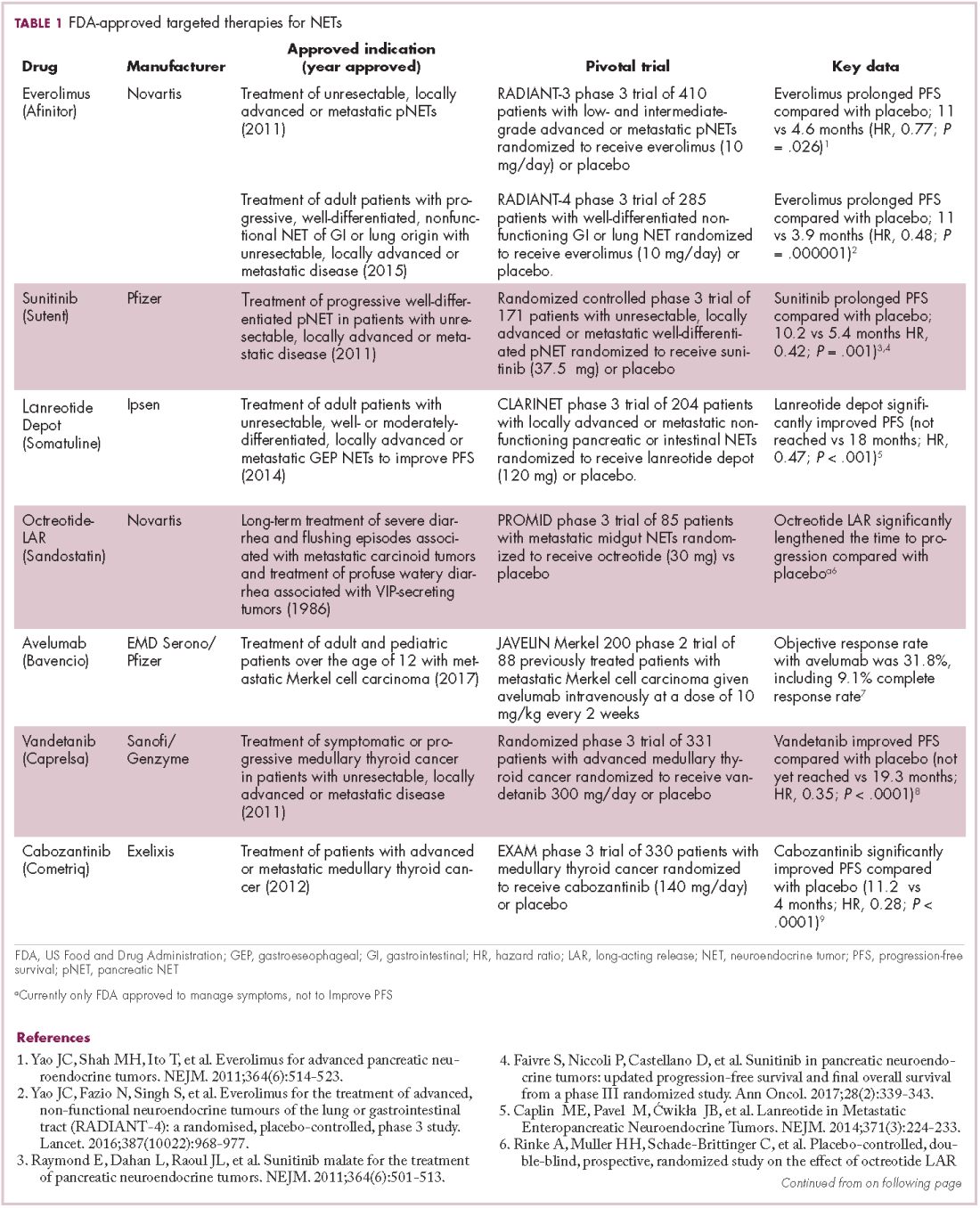
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).

Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
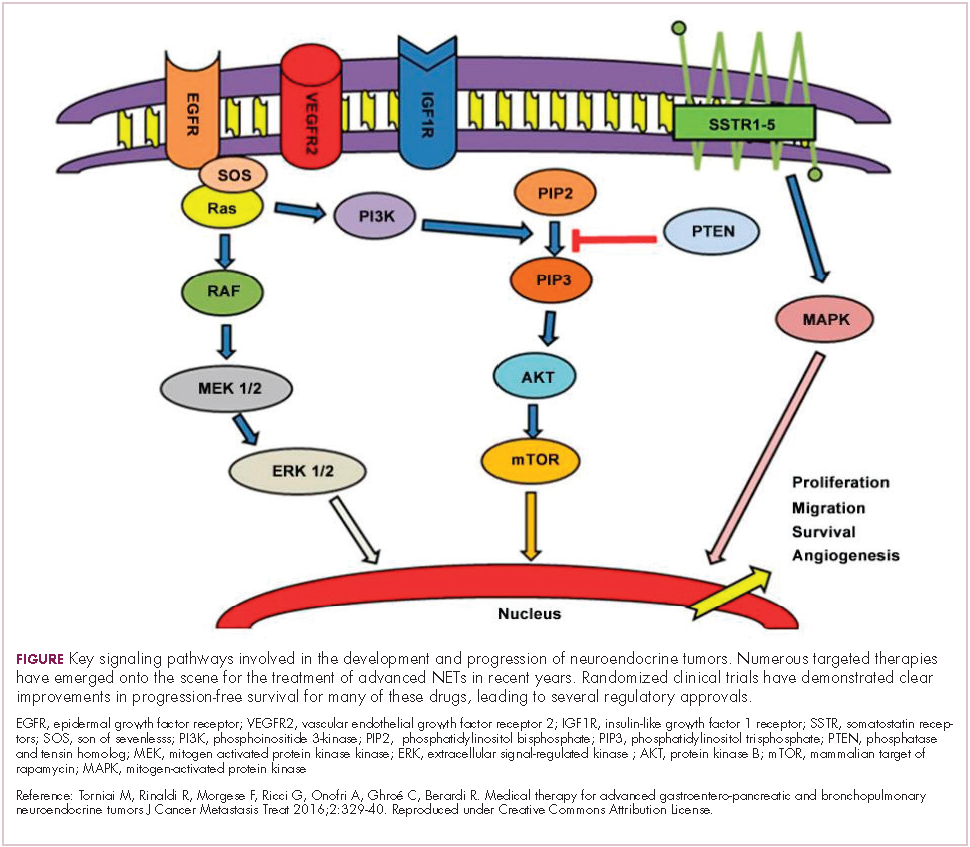
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
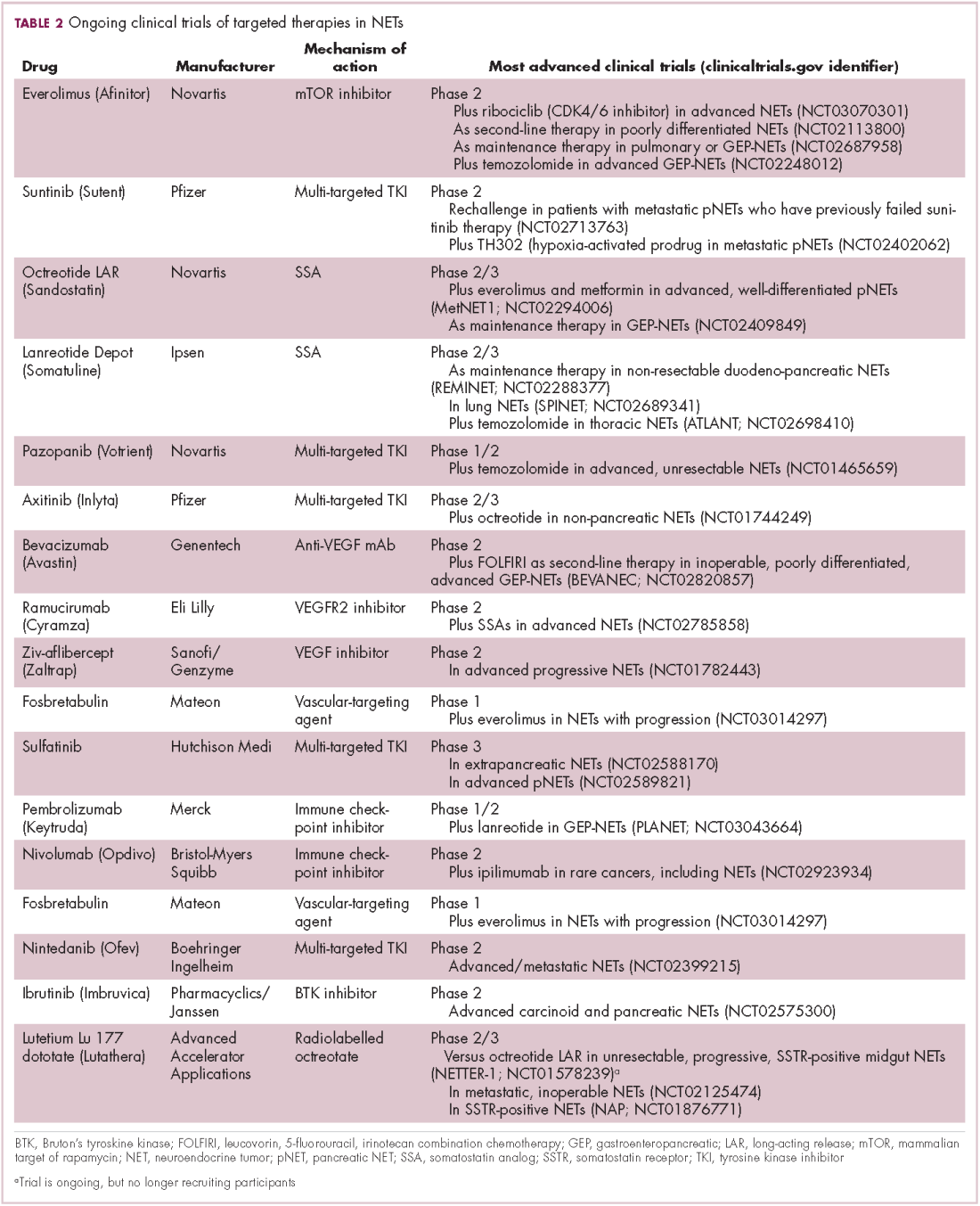
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29