Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
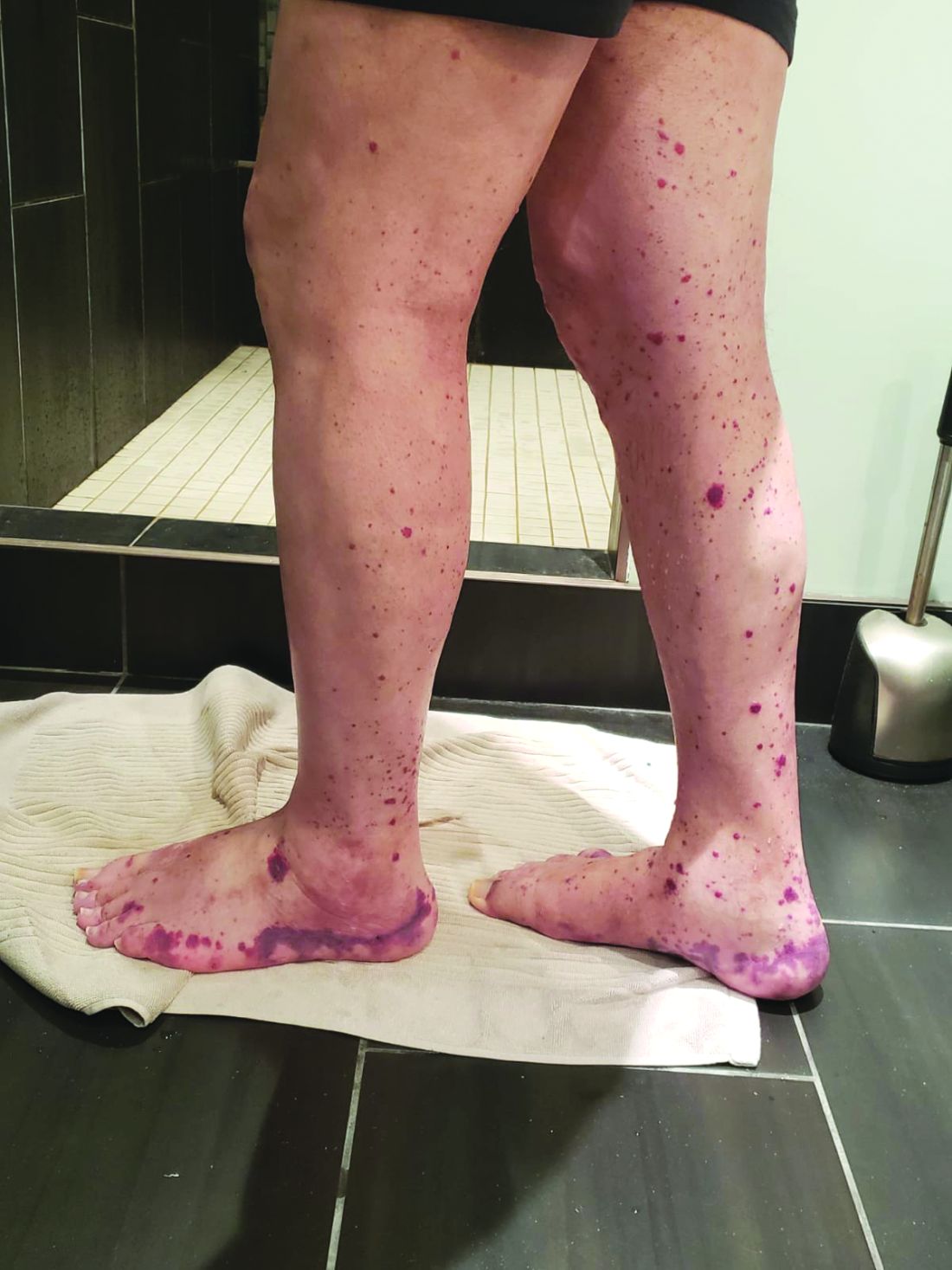 Courtesy Dr. Marcela Ferrada
Courtesy Dr. Marcela Ferrada
Leukocytoclastic vasculitis seen in legs and feet of a man with VEXAS.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”