Drs. Galve and Ferrando are from the Department of Dermatology, Hospital Clínic, University of Barcelona, Spain. Drs. Martín-Santiago, Clavero, Saus, Alfaro-Arenas, Pérez-Granero, and Balliu are from University Hospital Son Espases, Palma de Mallorca, Spain. Dr. Martín-Santiago is from the Department of Dermatology; Drs. Clavero and Balliu are from the Department of Pediatrics; Dr. Saus is from the Department of Pathology; and Drs. Alfaro-Arenas and Pérez-Granero are from the Department of Genetics.
The authors report no conflict of interest.
Correspondence: Javier Galve, MD, Department of Dermatology, Hospital Clínic, Villarroel 170, 08036 Barcelona, Spain (jgalveclinic@gmail.com).
Silvery hair is a characteristic finding of 3 rare autosomal recessive disorders: Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS). We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have one of these classic causative disorders. The patient was delivered at 35 weeks’ gestation with congenital hydrops fetalis associated with a chromosomal abnormality (46,XY,add[2],[p23]), hypothyroidism, hypoproteinemia, and hypogammaglobulinemia. Over the course of follow-up, spontaneous brown repigmentation of the silvery hair was noted. We concluded that the silvery hair was induced by hypoproteinemia secondary to congenital hydrops fetalis.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
References
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
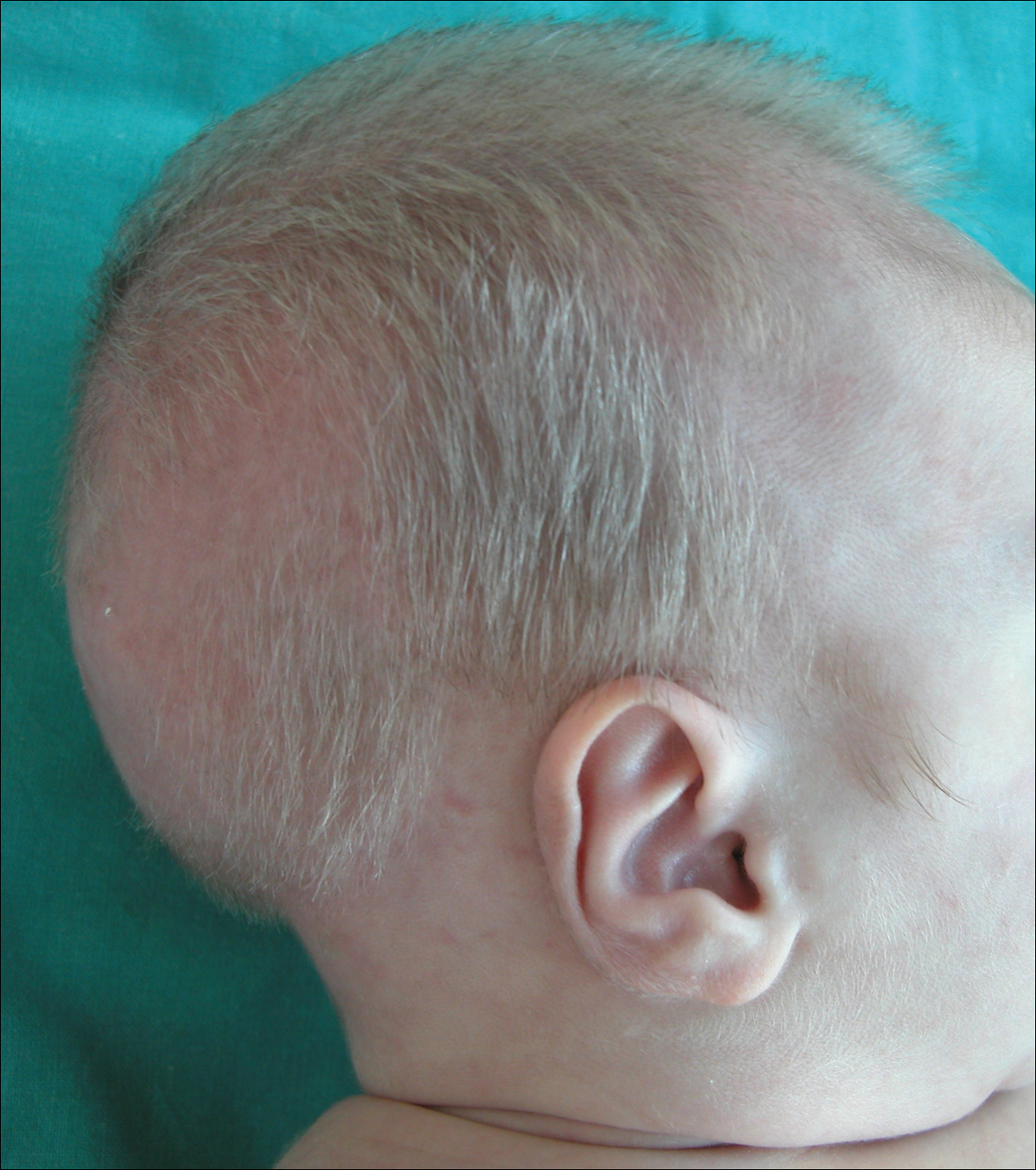
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Figure 1. A 2-month-old male infant with silvery scalp hair and generalized hypopigmentation of the skin.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
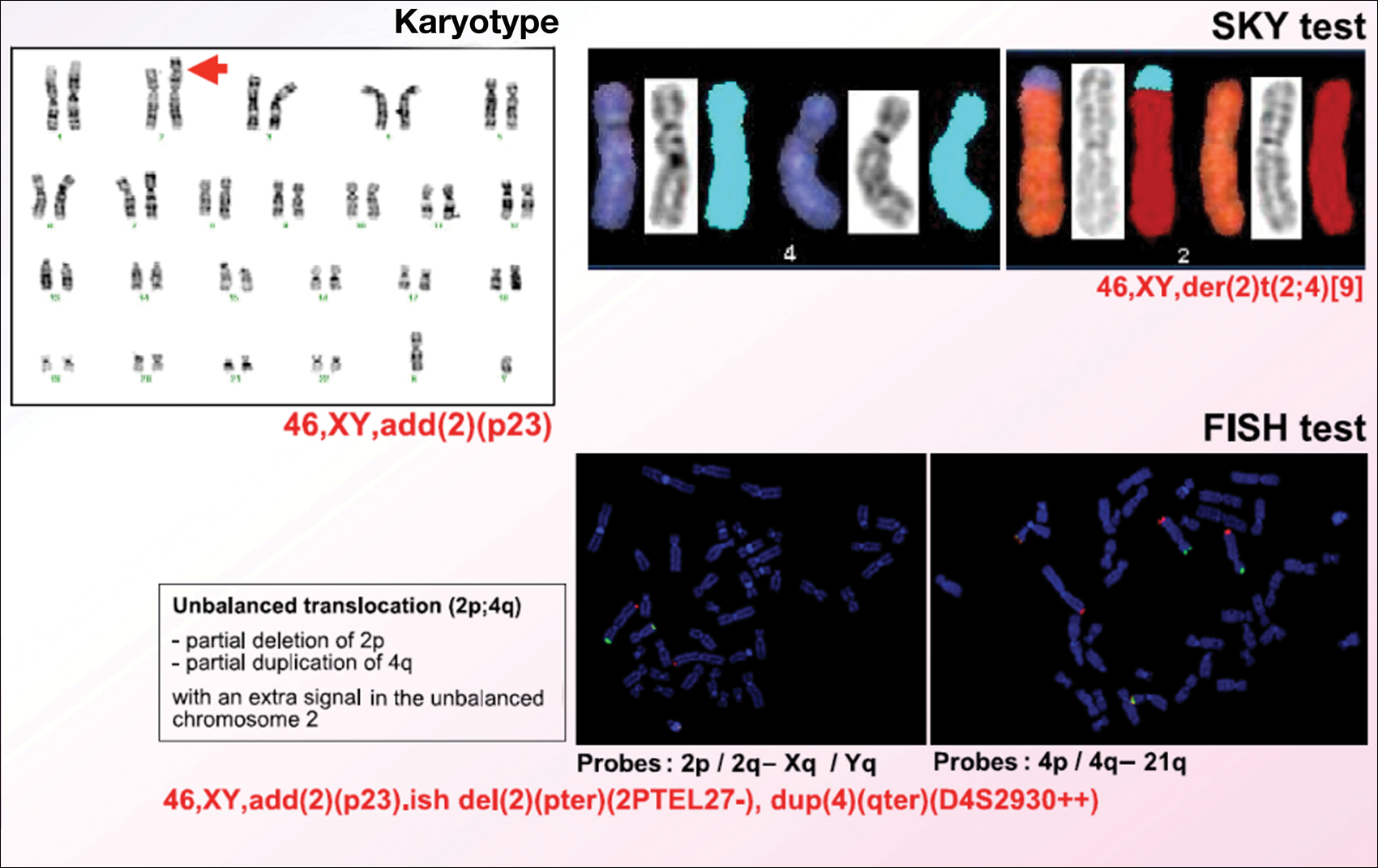
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
Figure 2. Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping (SKY) and fluorescence in situ hybridization (FISH) tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930 ]).
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
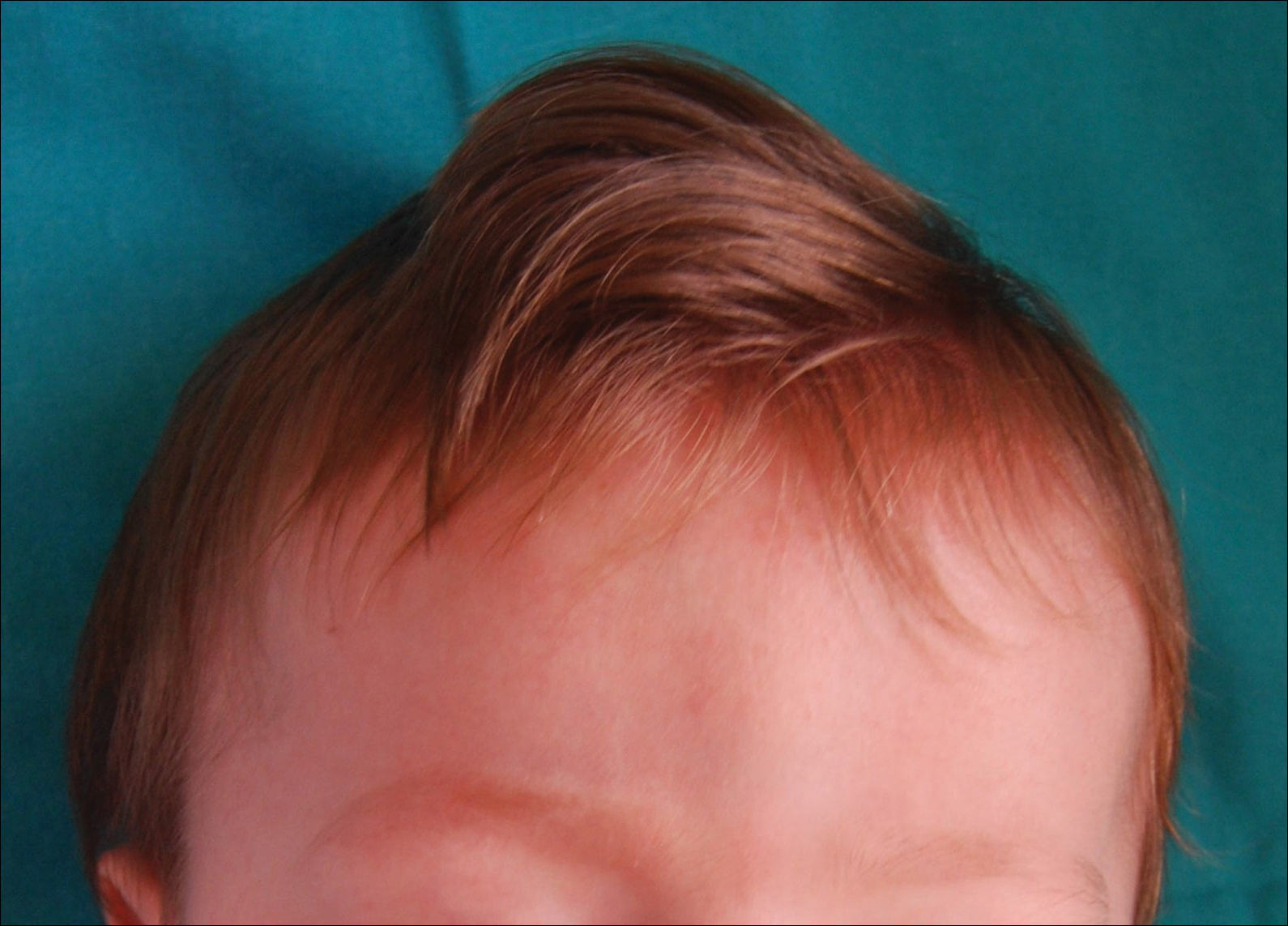
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Figure 3. Light microscopy of the hair showed small clumps of melanin pigment evenly distributed, predominantly in the medulla.
Figure 4. At 9 months of age, the patient showed spontaneous brown repigmentation of the silvery hair.
Comment
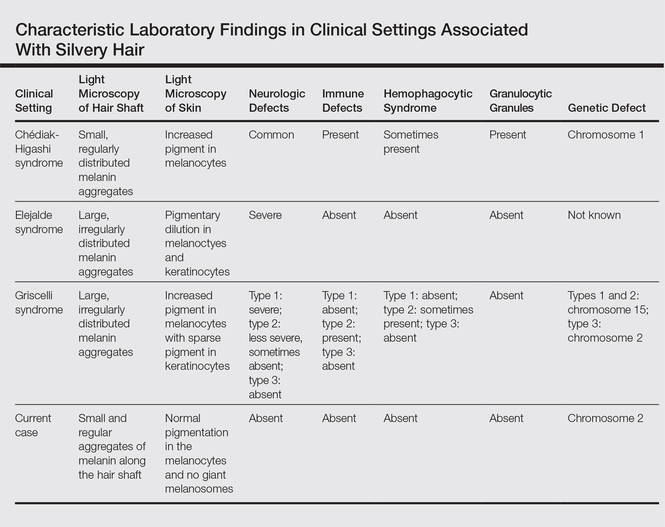
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.