Ms. Patel and Dr. Gilson are from the University of Texas Health Science Center, San Antonio. Ms. Patel is from the Long School of Medicine, and Dr. Gilson is from the Division of Dermatology. Dr. Gillooly is from the Department of Dermatology, Wright-Patterson Medical Center, Wright-Patterson Air Force Base, Ohio. Dr. Brown is from the Division of Dermatology, University of Texas Health, Austin.
The authors report no conflict of interest.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Air Force, Department of Defense, or the US Government.
A 67-year-old woman with a medical history of type 2 diabetes mellitus, unspecified leukocytosis, and anemia presented to the dermatology clinic with asymptomatic hemorrhagic bullae on the face, chest, and tongue, as well as a large, tender, tense, hemorrhagic bulla on the groin of 3 to 4 months’ duration. A review of systems was negative for fever, chills, night sweats, malaise, shortness of breath, and dyspnea on exertion. A complete blood cell count showed mild leukocytosis, anemia, and thrombocytopenia. Her creatinine level was slightly elevated. Chest computed tomography showed early pulmonary fibrosis and coronary artery calcification. An echocardiogram showed diastolic dysfunction with moderate left ventricle thickening. A serum and urine electrophoresis demonstrated elevated free λ light chains with an M-spike. A punch biopsy was performed.
The Diagnosis: Bullous Amyloidosis
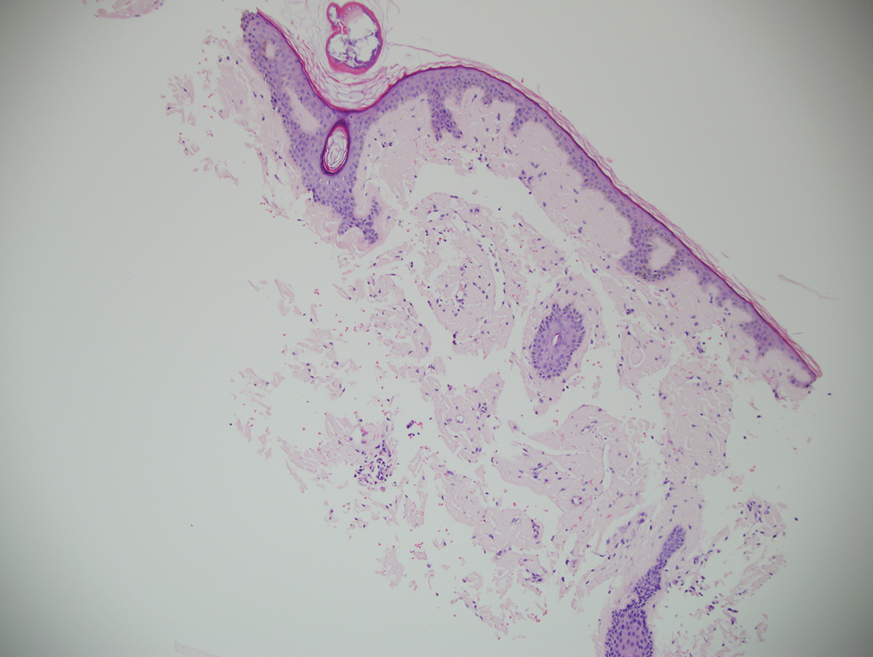
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Figure 1. Bullous amyloidosis. Histopathology demonstrated deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (H&E, original magnification ×10).
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
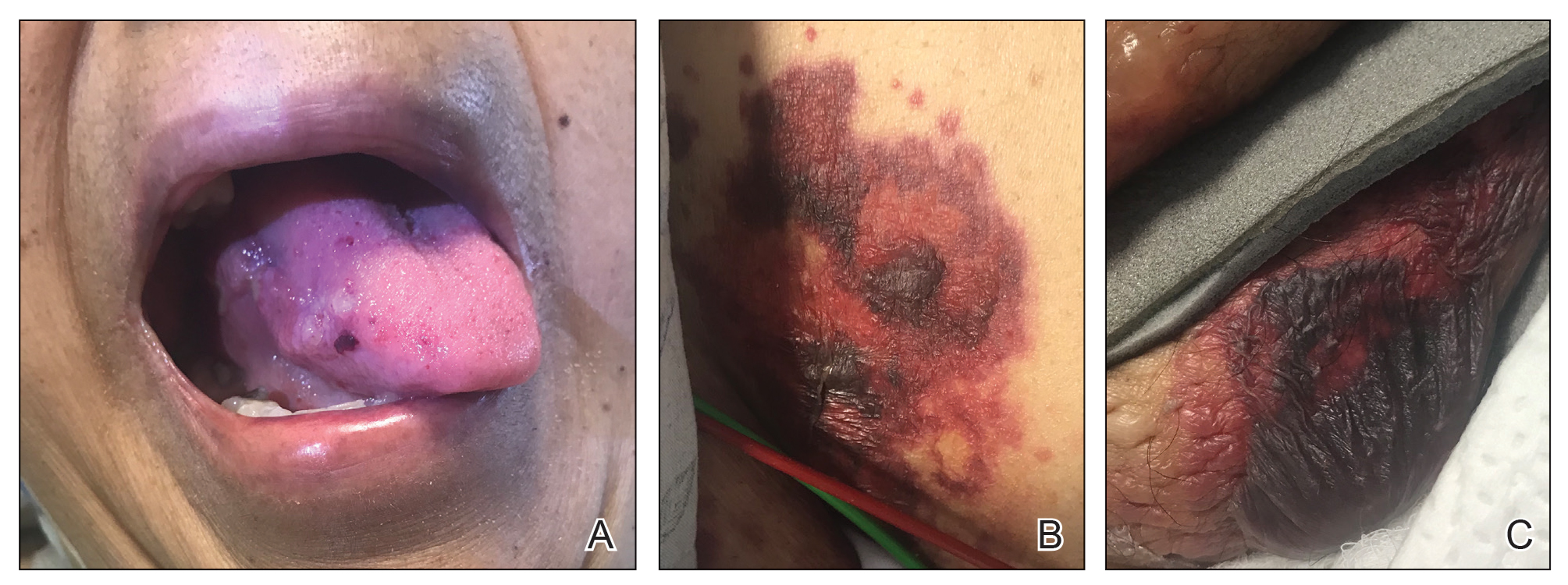
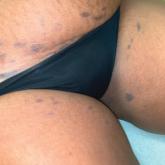
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
Figure 2. Bullous amyloidosis. A, Localized hemorrhagic bulla on the lateral tongue. B and C, Large hemorrhagic bullae on the groin.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.