
The Diagnosis: Primary Systemic Amyloidosis
Our patient presented with multiple firm waxy nodules on the mucosal surface of the lower lip. Excision biopsy showed thickening of blood vessel walls with abundant amorphous material that was consistent with amyloid. Further staining with Congo red demonstrated brick red amorphous material within the vessel walls on routine light microscopy (Figure 1), and crystal violet stain showed metachromasia (Figure 2). Fine needle aspiration of the abdominal fat-pad showed amyloid. The final diagnosis was primary systemic amyloidosis (PSA).
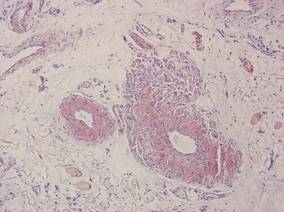 |
| Figure 1. Congo red staining showed a brick red appearance of amyloid within the vessel walls on routine light microscopy (original magnification ×200). |
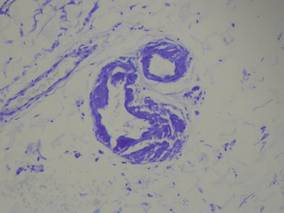 |
| Figure 2. Crystal violet staining around the blood vessels showed metachromasia (original magnification ×100). |
Amyloid is an ubiquitous fibrillar protein arranged in a cross-beta-pleated sheet that is confirmed with x-ray crystallography.1,2 More than 25 variants of amyloid have been identified.3 Pathologic deposition of amyloid-derived material results in a variable spectrum of clinical findings, collectively known as amyloidosis, with presentations ranging from nonspecific fever or fatigue to frank organ failure, depending on the organ involved. In PSA, immunoglobulin light chains are deposited throughout the body. Associated conditions include malignant or benign monoclonal gammopathy, multiple myeloma, Waldenström macroglobulinemia, malignant lymphoma, heavy chain disease, and chronic lymphocytic leukemia.2,4 The most commonly involved organ systems are the heart, lungs, liver, and kidneys. When patients present with unexplained heart failure, orthostatic hypotension, hepatomegaly, peripheral neuropathy, carpal tunnel syndrome, or renal insufficiency, amyloidosis should always be considered in the differential diagnosis.3
Cutaneous lesions of PSA tend to be vascular due to amyloid infiltration of blood vessel walls, manifesting as petechiae, purpura, ecchymoses, or nonhealing ulcers. Pinch purpura frequently are seen in the periorbital region after minor trauma and are recognized as a clinical indicator of PSA.2,4 Xerostomia from amyloid infiltrates in salivary glands is extremely common, and cases of amyloid in the eyes, bones, and thyroid gland have been reported.2 Macroglossia is seen in 12% to 40% of cases; coupled with xerostomia, it can lead to oropharyngeal dysphagia.2,5
Systemic amyloidosis can be further divided into primary (idiopathic or multiple myeloma associated) or secondary to chronic inflammatory conditions or infections; the key difference is the protein from which the abnormal amyloid is derived.1,4 The presence of cutaneous amyloidosis renders the need to rule out systemic disease because amyloidosis may be a purely localized or systemic process.4 Nodular amyloidosis is a localized form of amyloid that also has immunoglobulin light chain deposits and clinically appears exactly the same as PSA; however, the deposits are restricted to the skin.1,2,4,5
Characteristic biopsy findings in cutaneous amyloidosis include amorphous orange-red amyloid deposits on hematoxylin and eosin–stained sections. The gold standard for amyloid detection is apple green birefringence under polarized light with Congo red stain or electron microscopy.6,7 Other stains used to identify amyloid include crystal violet, methyl violet, periodic acid–Schiff, Sirius red, pagoda red, Dylon stain, and thioflavine T.1,2 Confirmation of systemic disease can be accomplished by fine needle aspiration of abdominal fat-pads or rectal mucosal biopsies.1,3,4 Biopsy of accessory salivary glands also has been reported to be very sensitive and specific.2
Treatment options remain limited; localized cutaneous disease may respond to topical corticosteroids, calcineurin inhibitors, or phototherapy. Primary systemic amyloidosis can be treated with a combination of steroids, melphalan, or colchicine often followed by autologous stem cell transplantation8; however, these regimens are not always curative and patients often have a poor prognosis.1