A healthy 17-year-old adolescent boy with an unremarkable medical history presented with an asymptomatic fixed rash on the abdomen, buttocks, and legs. The rash initially developed in a small area on the right leg 2 years prior and had slowly progressed. He was not currently taking any medications and did not participate in intense physical activity. Multiple biopsies had previously been performed by an outside physician, the most recent one demonstrating an interface and superficial perivascular lymphocytic infiltrate with extravasated red blood cells consistent with pigmented purpura. He did not respond to treatment with intralesional corticosteroids, high-potency topical steroids, or high-dose oral prednisone.
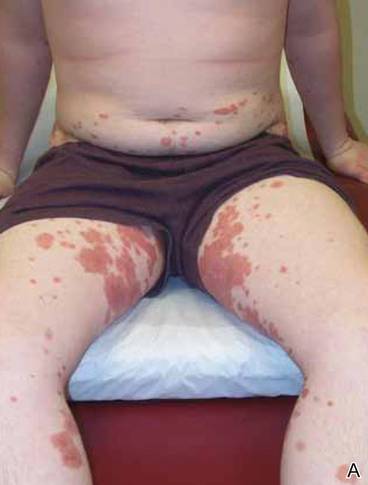
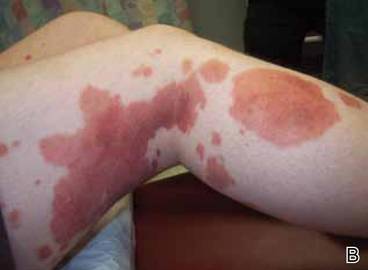
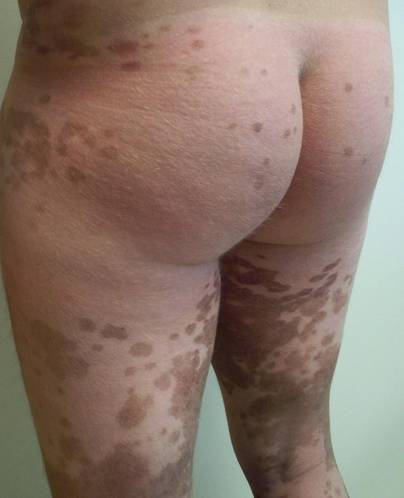
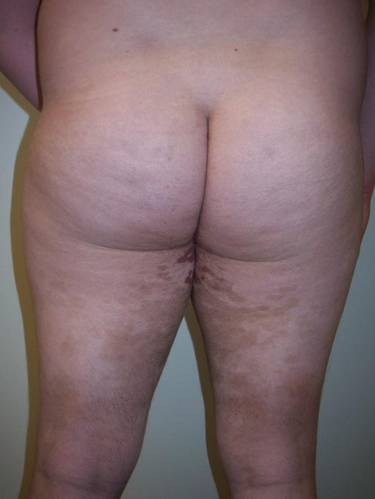
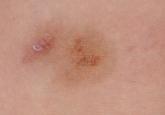
Clinical examination revealed multiple annular purpuric patches on the abdomen, buttocks, and legs that covered approximately 20% of the body surface area (Figure 1). Over several follow-up visits, a few of the lesions evolved from patches to thin plaques. There was no adenopathy or hepatosplenomegaly. Three additional biopsies taken over the next 4 months demonstrated a mixture of small mature lymphocytes with some atypical lymphocytes in the dermis and epidermis exhibiting diminished CD7 staining and lymphocytes lining up at the dermoepidermal junction. T-cell receptor g gene rearrangements demonstrated the same clonal population in all 3 specimens. The patient was diagnosed with stage IB mycosis fungoides (MF) of the pigmented purpura–like variant. Marked improvement of the lesions was noted after 6 weeks of psoralen plus UVA therapy 3 times weekly (Figure 2). Treatment was continued for 6 months but was discontinued due to the international shortage of methoxsalen. Two months after discontinuation, most of the lesions had completely resolved (Figure 3).
Figure 1. Multiple annular purpuric patches on the abdomen, anterior thighs (A), and right leg (B). |
Mycoses fungoides is a rare cutaneous lymphoma that affects approximately 2000 patients in the United States.1 Only 5% of all cases are known to occur in the first 2 decades of life,2 and even fewer cases pre-sent with pigmented purpura, usually of the lichenoid variant.3 Although the patches and plaques of MF can masquerade as many other dermatoses (eg, dermatophytosis, psoriasis, dermatitis), there have been few reports of patients presenting with lesions with the clinical appearance of pigmented purpuric dermatosis (PPD).4 As with the many cases of early MF, which are histologically indistinguishable from dermatitis, the pigmented purpura–like variant of MF initially may have the histologic appearance of pigmented purpura and generally evolves to the histologic appearance of MF over time.
Similar to our case, there have been reports of clinical and histologic diagnosis of PPD preceding the histologic diagnosis of MF. In a small cohort study of 3 young men, Barnhill and Braverman5 first demonstrated the progression of PPD to MF over a 12-year period. The age of onset ranged from 14 to 30 years, with a mean age of 24.3 years. Biopsies in all 3 patients were consistent with PPD for many years prior to the diagnosis of MF, with an average length of time to diagnosis of 8.4 years. Atypical from most cases of PPD, the patients in this study demonstrated extensive involvement of the trunk, arms, and legs.5 It has been suggested that atypical PPD is a variant of PPD that evolves into MF over many years; however, we believe that PPD is a variant of MF, similar to the way an indolent dermatitis may evolve to classical MF over time. If characterized by a T-cell clone, this period preceding the diagnosis of cutaneous T-cell lymphoma could be characterized as a cutaneous T-cell lymphoid dyscrasia.
Guitart and Magro6 noted multiple chronic conditions that are associated with T-cell clones, including PPD. These conditions occurred without a known trigger, were unresponsive to topical therapies, and often did not meet diagnostic criteria for MF. The investigators felt the criteria that may indicate a cutaneous T-cell lymphoid dyscrasia include widespread distribution, lymphocytic infiltrate, diminished CD7 and CD62L expression, and clonality. Lymphocytes may be small without notable atypia.6
In a study of 43 patients with PPD, Magro et al3 found monoclonality and diminished CD7 expression in 18 participants, correlating with large surface area involvement. Approximately 40% of patients had histologic findings consistent with MF, suggesting that T-cell gene rearrangement studies should be obtained for prognostic evaluation in patients with widespread disease.3
| Figure 2. Marked improvement of the lesions was noted after 6 weeks of psoralen plus UVA therapy 3 times weekly. | Figure 3. After 6 months of psoralen plus UVA therapy, most of the lesions had completely resolved. |
To facilitate proper patient care, histopathology and molecular markers should be evaluated in conjunction with the clinical picture. A considerable increase in the size of the body surface area affected by purpuric patches combined with the presence of poikilodermatous changes and pruritus as well as disease lasting longer than 1 year should prompt an increased clinical suspicion of MF in patients with PPD.4,5 Histologically, the presence of Pautrier microabscesses, large cerebriform lymphocytes, and intraepidermal lymphocytic atypia extending beyond the dermis also would support a diagnosis of MF.3 Given the morphologic appearance and distribution of the lesions in our patient combined with epidermotropism, diminished CD7 expression, and monoclonality seen on pathology, we favored a diagnosis of MF. It would not be unreasonable to call this clonal variant of PPD a T-cell lymphoid dyscrasia. We appreciate that both PPD and MF will respond to phototherapy.7