Bone Marrow Aspiration
Surprisingly, the FDA has not moved to regulate the point-of-care use of bone marrow aspirate or platelet-rich plasma and has labeled these as “not HCTPs.” The stem cell concentration of bone marrow aspirate is technique-dependent, declines with age, and has been found to be an important factor for clinical benefit.31 While it is possible to aspirate from multiple sites, posterior iliac crest harvest produces the highest stem cell yield.32-34 Hernigou and colleagues35-36 have outlined safe zones for trocar placement and illustrated that strong aspiration with small-volume syringes, 10-mL syringes, optimizes stem cell harvest. Additionally, studies by Hernigou and colleagues31,37-38 involving tibial nonunion, avascular necrosis of the femur, and augmentation of rotator cuff repair are guideposts to clinicians utilizing bone marrow aspirate.
Amniotic Stem Cell Technologies and Adipose-Derived Stem Cells
While some argue that there is regulatory confusion around amniotic/placental-derived tissues and adipose-derived products, the FDA has clearly established precedent establishing these as products requiring Section 351 development.26-29 Companies are marketing products derived from perinatal byproducts, yet there are multiple FDA letters suggesting that these are not products regulated solely under PHS Act 361 because they do not meet the criteria of homologous use and are not autologous.28-29 Use of these products places risk upon the clinician and the patient. Some argue that adipose-derived stem cell products are 361 products. While the FDA has approved devices for the mechanical processing of lipoaspirate, they have established precedent suggesting that they consider orthopedic applications nonhomologous and any processing that “alters the original relevant characteristics of adipose tissue relating to the tissue’s utility for reconstruction, repair, or replacement” as more than minimal manipulation.26,27 The FDA originally planned an open forum for discussion with clinicians and industry for April 2016. This open forum was delayed due to the volume of interest, and a workshop has been planned for Fall 2016.
Future Regulation of Stem Cell Technologies
While many countries have mirrored the FDA with tight regulatory mechanisms, a few countries have established modern regulatory mechanisms aimed at the promotion of conscientious development, including South Korea, Japan, and England. For example, in 2014 Japan labeled stem cell technologies as “regenerative medicine products,” setting them apart from pharmaceuticals, and implemented a new approval system allowing early observed commercialization with reimbursement after less stringent safety and efficacy milestones.22The observed commercialization lowers time and financial hurdles for development while still requiring the proof of the technology’s worth. Countries that have effected change have positioned themselves to be pioneers in this emerging field.
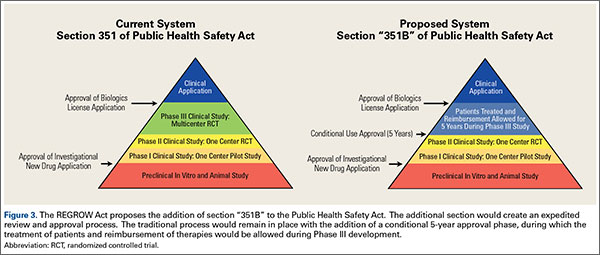
In March 2016, the Reliable and Effective Growth for Regenerative Health Options that Improve Wellness (REGROW) Act of 2016 (S. 2689 / H.R. 4762) was introduced into the United States Congress. This bipartisan, bicameral legislation was introduced, read twice, and referred to subcommittee. Its goal is to reduce barriers and accelerate development of biologic therapies while keeping the frame work set forth under Sections 351 and 361 of the PHS Act.39 Similar to the pathway in Japan, the REGROW Act would establish a conditional approval pathway that would ensure products are safe and effective while also evolving the regulatory pathway towards progress (Figure 3). Development would still require an IND application after preclinical animal study. However, after safety was established with human Phase I data and preliminary evidence of efficacy with Phase II data, patients could be treated with the investigational therapies and reimbursement collected for a limited period of time (5 years) prior to a large Phase III human clinical trial. Patients treated with the new therapy would be monitored closely. All results would be reported to the FDA in a BLA. This change in legislation would lower but not remove regulatory hurdles necessary for development.
Conclusion
The future of stem cell treatments hinges upon the creation of new favorable regulatory mechanisms that will promote clinical application while ensuring that safety and efficacy milestones are reached. Clinical researchers require freedom to develop these technologies while protecting patients and ensuring the validity of treatments. The coordination of research and regulatory affairs on a global level is necessary focusing on the harmonization of guidelines, regulations, and mechanisms for simultaneous adoption in different countries. The global orthopedic community has made strides regarding the science of stem cell technologies; it is time for us to initiate progressive change regarding regulation so that we can determine what is effective clinically.