Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
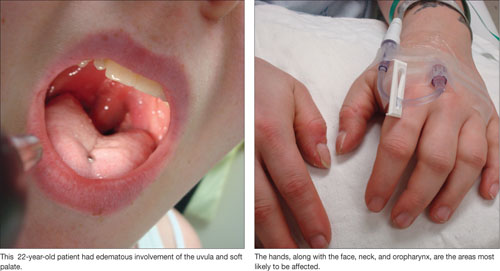
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20