SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel recommended against approval of a low-dose formulation of paroxetine mesylate as a treatment for menopausal vasomotor symptoms, citing minimal benefits over placebo in clinical trials and the potential risks associated with treatment.
At the March 4 meeting, the FDA’s Reproductive Health Drugs Advisory Committee voted 10 to 4 that the overall risk-benefit profile of paroxetine did not support approval of the indication proposed by the manufacturer, Noven Therapeutics, as a treatment for moderate to severe vasomotor symptoms associated with menopause, at a dose of 7.5 mg once a day at bedtime.
The 7.5-mg dose is lower than the doses approved for psychiatric indications of paroxetine, which range from an initial dose of 10 mg to a maximum of 60 mg per day, depending on the indication.
Currently, paroxetine, a selective serotonin reuptake inhibitor (SSRI), is used off-label to treat vasomotor symptoms (VMS), and if approved, this could be the first nonhormonal treatment for VMS associated with menopause. Earlier in the day, the same panel voted against recommending approval for the antiepileptic drug gabapentin, another drug used off-label for VMS, for the same indication, citing the drug’s modest effects on VMS and its potential risks. In studies of both drugs, there was a marked placebo effect, which complicated the evaluations of the effectiveness.
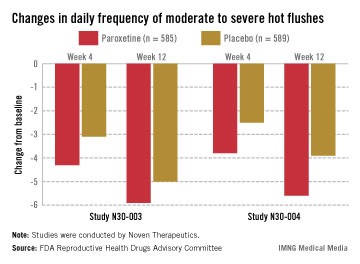
In two pivotal studies conducted in the United States, 1,180 women (mean age, 54-55 years) with natural or surgical menopause who had at least 7 hot flashes per day or at least 50 per week were randomized to receive paroxetine or placebo, after a 12-week run-in period. Electronic diaries were used to document hot flashes. The primary endpoints were the mean change from baseline in hot flash frequency, and the mean change from baseline in hot flash severity, at weeks 4 and 12.
Those on paroxetine had statistically significant reductions in the daily number of hot flashes from baseline, compared with those on placebo at weeks 4 and 12 in both studies, and at week 24 in one of the two studies, which was a 24-week study. The reductions in the daily severity of hot flashes also were statistically significant at week 4 in both studies, and at week 12 in one study but not in the other study, according to the FDA.
However, the women on placebo had notable responses, not unexpected for a nonhormonal treatment, and the difference in the median number of hot flashes per day between the two groups was less than 2: In one study, the median number of hot flashes a day dropped by 4.29 by week 4 and 5.93 at week 12 among those on paroxetine, versus 3.13 and 5, respectively, among those on placebo. In the other study, the median number of hot flashes a day dropped by 3.79 at week 4 and 5.57 by week 12 among those on paroxetine, versus 2.5 and 3.86, respectively, among those on placebo.
The safety profile in the studies was consistent with the known safety profile of paroxetine, with no new or unexpected findings, according to Noven. Fatigue, nausea, and dizziness were the most common side effects in people treated with paroxetine, but they occurred mostly in the first 4 weeks of treatment and were mild to moderate Serious adverse events were higher among those on paroxetine (2.2% vs. 1.4%). In the 24-week study, there were three cases of suicidal ideation and one suicide attempt among those on paroxetine, all after 12 weeks of treatment. The one death in the studies was in a woman on paroxetine, but the death not considered related to the drug, according to the company.
Paroxetine was first approved in 1992 as paroxetine hydrochloride. Paroxetine mesylate was first approved in 2003 and is marketed as Pexeva; it is approved for major depressive disorder, obsessive compulsive disorder, panic disorder, and generalized anxiety disorder. As with all antidepressants, it has a boxed warning about the risk of suicidality and serotonin syndrome associated with treatment.
Noven refers to low-dose paroxetine as LDMP, for low-dose mesylate salt of paroxetine.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
emechcatie@frontlinemedcom.com