Practice recommendations
- Echocardiography is useful for screening high-risk patients (SOR: A).
- The New York Heart Association classification of dyspnea has been modified by the World Health Organization to categorize pulmonary hypertension by the severity of symptoms, which, unlike pulmonary arterial pressure, correlates well with survival (SOR: A).
- Calcium channel blockers are useful only for patients who respond to vasodilator testing in a cardiac catheterization laboratory (SOR: A).
- Therapeutic modalities now include parenteral prostanoids, oral endothelin receptor antagonists, PDE5 inhibitors, and lung transplantation (SOR:A; for PDE5 inhibitors, SOR: B)
- Early referral to expert centers is crucial to patient survival (SOR: B).
Recent progress in understanding the pathobiology of pulmonary arterial hypertension (PAH) has been tremendous, and treatment options have multiplied to include prostanoids, endothelin antagonists, phosphodiesterase-5 inhibitors, anticoagulants, and surgical options such as lung transplantation and atrial septostomy.
Although idiopathic pulmonary arterial hypertension, formerly called “primary,” is rare, other forms of PAH and associated cor pulmonale are more prevalent than conventionally believed. It is a life-threatening disease best managed within a diagnostic framework such as the one reviewed here with a treatment algorithm and recommendations from evidence-based guidelines.
Patients most likely to experience pulmonary arterial hypertension
Pulmonary arterial hypertension may be idiopathic and sporadic (IPAH), familial (FPAH), or associated with (APAH) connective tissue diseases, congenital systemic to pulmonary shunts, portal hypertension, HIV, drugs including anorexigens or cocaine, and other disorders ( Table 1 ).1
Annually, 1 to 2 cases of IPAH occur per million population.2 The mean age at diagnosis is 36 years, and women are affected more often than men by a ratio of 1.7–3.5:1. This female predominance has also been noted in PAH associated with scleroderma,3 congenital heart disease,4 and anorexigen-induced PAH.5 The incidence among users of anorexigens such as fenfluramine, dexfenfluramine, and aminorex is estimated to be 25 to 50 per million per year.2
The prevalence of portopulmonary hypertension is about 0.73% in cirrhosis.6 In scleroderma, the incidence is between 6% to 60%,7,8 while in systemic lupus erythematosus (SLE) it is reported to be 4% to 14%.9,10 In one study, 21% of rheumatoid arthritis patients without underlying cardiopulmonary disease had mild pulmonary hypertension (PH).11 PAH occurs in about 0.5% of patients with HIV infection.12
Included in the “others” group are hemoglobinopathies such as sickle cell anemia. This classification does not include PH due to end-stage renal disease, a recently described entity in patients with arteriovenous fistulae that portends a poorer prognosis.13 PH was present in a surprising 40% of hemodialysis patients.
TABLE 1
The 2003 Venice clinical classification of pulmonary hypertension*
|
| *Classification does not include pulmonary hypertension due to end-stage renal disease.13 |
Ortner’s syndrome

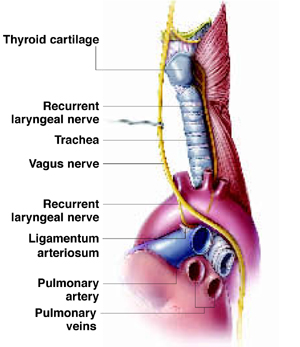
Ortner’s syndrome was first described in 1897. The commonest cause has been thought to be mitral valve stenosis or prolapse leading to left atrial dilatation that compresses the recurrent laryngeal nerve as it hooks around the aorta next to the ligamentum arteriosum and courses up in the groove between the trachea and esophagus. However, other vascular structures can also compress this nerve, including a thoracic aortic aneurysm, or a dilated pulmonary artery from pulmonary arterial hypertension.
Clinical presentation
Pulmonary arterial hypertension manifests the following symptoms and signs:
Symptoms
- Progressive onset of exertional dyspnea (60%)14
- Chest pain or discomfort (17%)
- Palpitations (5%)
- Dizziness and light-headedness. There may be a history of near-syncope or syncope (13%)
- Fatigue (19%)
- Ortner’s syndrome: hoarseness from compression of left recurrent laryngeal nerve by enlarged pulmonary artery (<1%) (See Ortner’s syndrome)
- Raynaud’s phenomenon (10%)
Signs
- Loud P2 (93%)
- Tricuspid regurgitation murmur (40%)
- Right ventricular heave
- Jugular venous distention with a prominent “a” wave
- Graham Steell’s murmur: diastolic pulmonary regurgitation murmur best heard at upper left sternal border (13%)
- Signs of right heart failure including S3 gallop, “v” wave in central venous pressure tracing, hepatojugular reflux, peripheral edema, and ascites
- Cutaneous telangiectasia.