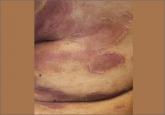
Diagnosis of chromoblastomycosis is often delayed because of the slow-growing nature of the plaques, its few initial symptoms, and the rarity of the condition in nonendemic regions.1,2 A skin scraping may demonstrate hyphae or spores. On histology, clusters of brown spores (“copper pennies”) are pathognomonic.5 A biopsy and tissue culture establish the diagnosis. The most frequent organisms identified include Phialophora, Fonsecaea, and Cladosporium.1
More than one type of treatment may be necessary
Antifungals are the mainstay of therapy for chromoblastomycosis, but due to frequent recurrences, multiple types of treatment are often necessary. Chromoblastomycosis is often extremely difficult to treat; frequent recurrences may require multimodal therapy.6,7 Treatment options include pharmacotherapy, surgery, and destructive therapy.
Systemic antifungals are the mainstay of therapy because topical antifungals do not penetrate deep enough in the skin. Several regimens have been presented in the literature. A common starting point is oral terbinafine8 250 to 500 mg/d and itraconazole9 200 mg to 400 mg/d for 6 to 12 months. Another medication used to treat chromoblastomycosis is intralesional or intravenous amphotericin B dosed variably with or without 5-flucytosine until the plaques clear.10 Based on small trials, treatment efficacy with antifungals ranges from 70% to 90%.6,7 Treatment often is limited by medication interactions and adverse effects.
Surgical therapy. A solitary plaque or smaller plaques may be removed with good outcomes. However, plaques may be so large that surgical removal is impractical or disfiguring.5,11